如何计算甲基化转化率?
BS转化后需要加入一段已知lambdaDNA序列。
比较sam文件中CT转换情况和lambdaDNA的ref原始碱基进行比较。
工具:MethylExtractBSCR.pl
-
下载链接:
https://bioinfo2.ugr.es/MethylExtract/ -
工具参数:
perl MethylExtractBSCR.pl
seqFile = <sequence file>
inFile = <alignments input file>
flagW = <Watson FLAGs>
flagC = <Crick FLAGs>
-
单端测序:
0代表单端测序,16代表这个序列比对到参考序列的负链上。
MethylExtractBSCR.pl seqFile=lambdaDNA.fa inFile=input.lambda.sam flagW=0 flagC=16 -
双端测序:
99代表双端测序、和参考序列完全匹配、比对到正链、first in pair
147代表双端测序、和参考序列完全匹配、比对到负链、second in pair
83代表双端测序、和参考序列完全匹配、比对到负链、first in pair
163代表双端测序、和参考序列完全匹配、比对到正链、second in pair
MethylExtractBSCR.pl seqFile=lambdaDNA.fa inFile=input.lambda.sam flagW=99,147 flagC=83,163
统计方法:二项分布和FDR校正
分析用文件:bismark_cov
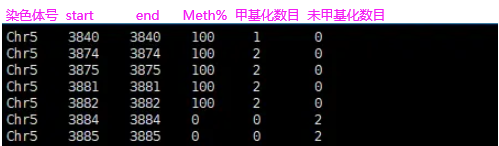
在转化率为A的基础上,N个reads中被检测到X个甲基化reads是否可靠。需要用二项分布来证明,FDR来校正。
p = binom.test(x,N,A)$p.value
fdr = p.adjust(p,"fdr")
FDR校正后,只保留FDR<0.01的甲基化位点。再过滤read数目<5的位点。
最后
以上就是清秀果汁最近收集整理的关于甲基化转化率计算的全部内容,更多相关甲基化转化率计算内容请搜索靠谱客的其他文章。
本图文内容来源于网友提供,作为学习参考使用,或来自网络收集整理,版权属于原作者所有。








发表评论 取消回复